{kind=link}
{kind=link}
{kind=link}
Two browser tools for Boltz-2 structure & binding prediction —
v1 generates local-CLI YAML for ternary complexes · v2.0 builds & submits hosted-API jobs for any complex.





| 🧪 BoltzYML v1 | 🚀 BoltzYML v2.0 |
|---|---|
| Open v1 web app → | Open v2.0 web app → |
Ternary Protein 1 + Ligand + Protein 2 preprocessing. Drop two CIFs → emits a ready-to-run
Boltz CLI YAML (version: 1) with pocket constraints and an affinity block, for
local boltz predict. Also shipped as a PyPI CLI.
→ v1 details below |
Any binary / ternary / N-body complex (protein · ligand · DNA · RNA). Builds the
hosted Boltz-2 API payload, auto-cleans template CIFs in the browser, and
submits with your own API key — then polls jobs and downloads results.
→ v2.0 details below |
v1 details · v1 CLI · How v1 works · v2.0 details · How to use v2.0 · Get an API key
index.html+ PyPI CLI — the original tool. Live at https://ayushmania2002.github.io/boltzyml/. Generates the open-source Boltz CLI YAML (version: 1) for runningboltz predicton your own machine.
Use this tool only if you want to study how the interaction between two proteins changes in the presence of a ligand.
That is the one scenario it is built for: a small molecule sits in a pocket of Protein 1, and you want Boltz-2 to predict how Protein 1 and Protein 2 dock together while that ligand is bound (or absent, as a control). Typical use cases:
- ABA signaling: PYR/PYL/RCAR receptor + ABA + PP2C phosphatase
- Allosteric drug screens where a ligand is expected to enable or block partner binding
- Any ternary system where the ligand sits on Protein 1 and you care about the Protein 1 ↔ Protein 2 interface
If your problem is just protein–protein docking, just protein–ligand docking, or anything that is not a ternary Protein 1 + Ligand + Protein 2 complex, BoltzYML will not help — write the YAML directly or use the Boltz-2 examples.
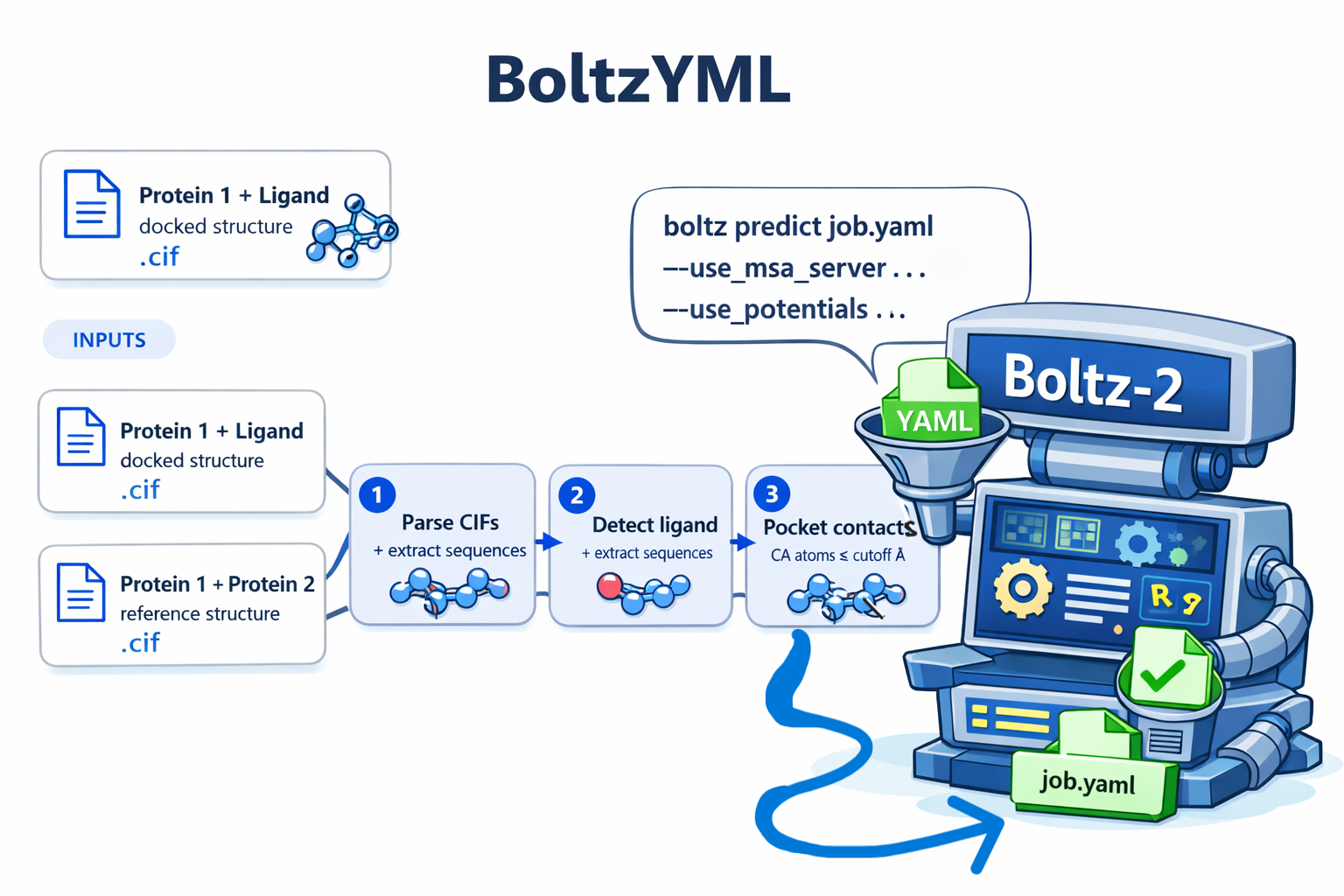
You hand BoltzYML two CIF files:
| Input | Contents | Role |
|---|---|---|
protein1_ligand.cif |
Protein 1 bound to the ligand (docked structure, e.g. AlphaFill, Glide, AutoDock) | Source of Protein 1 sequence + ligand identity + pocket residues |
protein1_protein2.cif |
Protein 1 together with Protein 2 (e.g. AlphaFold3 prediction) | Source of Protein 2 sequence |
BoltzYML then runs entirely in your browser (or via the CLI) and produces a single Boltz-2 v1 YAML with:
- Protein 1 sequence on chain
A - The ligand on chain
B(with its PDB CCD code) - Protein 2 sequence on chain
C - A
pocketconstraint listing the Protein 1 CA atoms within a cutoff of the ligand - An
affinityproperty block targeting the ligand
That YAML is then fed straight into the Boltz-2 CLI.
Open https://ayushmania2002.github.io/boltzyml/, drop in the two CIFs, click Generate YAML, download the file.
Everything happens in your browser — no uploads, no server, no telemetry. Works on any modern browser, no installation required.
Install from PyPI:
pip install boltzymlThen run:
boltzyml \
--ligand PYL2_ABA.cif \
--complex PYL2_PP2C30.cif \
--output PYL2_ABA_PP2C30.yamlPure Python 3.10+, zero runtime dependencies (no gemmi, no numpy, no pyyaml required).
You can also clone the repo and run it as a module without installing:
git clone https://github.com/Ayushmania2002/boltzyml.git
cd boltzyml
pip install -e .
boltzyml --ligand A.cif --complex B.cif -o out.yaml| Flag | Description | Default |
|---|---|---|
--ligand |
CIF with Protein 1 + Ligand | required |
--complex |
CIF with Protein 1 + Protein 2 | required |
-o, --output |
Output YAML path | {ligand-stem}__{complex-stem}.yaml |
--job-name |
Job name used for the default output filename | derived |
--cutoff |
CA-to-ligand distance cutoff in Å | 6.0 |
--ccd |
Override the ligand CCD code (used verbatim in the YAML's ccd: field) |
auto-detect |
--no-affinity |
Skip the affinity prediction block | off |
--no-pocket |
Skip the pocket constraints block | off |
--no-force |
Emit force: false on the pocket block |
off |
-v, --verbose |
Print detected chains, ligand, and contacts | off |
- Parse both CIFs. The parser handles two layouts seen in the wild: single-line atom records (AlphaFold3 style, 18 fields per line) and two-line records (some AlphaFill outputs, 21 fields split 17 + 4).
- Identify the ligand. The first non-water HETATM whose comp ID is not a standard amino acid is taken as the ligand. If the CIF labels it generically (
LIG,UNL,UNK), the tool warns and asks for a CCD override. - Assign chains. Protein 1 = the longest protein chain in the ligand CIF. The corresponding chain in the complex CIF is matched by 5-mer overlap similarity; the remaining chain is Protein 2.
- Compute pocket contacts. CA atoms of Protein 1 within
--cutoffÅ of any ligand atom are emitted as[A, residue_number]entries, using the CIF'sauth_seq_idso the numbers match Boltz-2's own residue numbering. - Emit YAML. A deterministic Boltz-2 v1 YAML is written out, in the field order Boltz-2 expects.
Why CA-only at 6 Å? CA-to-ligand at 6 Å approximates all-atom contacts at ~4 Å, which is the right scale for the soft pocket constraint Boltz-2 applies as an inference-time potential.
version: 1
sequences:
- protein:
id: A
sequence: MEAHVERALREGLTEEERAALEPAVMAHHTFPPSTTTATTAAATCTSLVTQRVAAPVRAVWPIVRSFGNPQRYKHFVRTCALAAGDGASVGSVREVTVVSGLPASTSTERLEMLDDDRHIISFRVVGGQHRLRNYRSVTSVTEFQPPAAGPAPAPPYCVVVESYVVDVPDGNTAEDTRMFTDTVVKLNLQKLAAVAEDSSSASRRRD
- ligand:
id: B
ccd: A8S
- protein:
id: C
sequence: MAEICCEVVAGSSSEGKGPECDTGSRAARRRR...
constraints:
- pocket:
binder: B
contacts:
- [A, 76] # PHE76
- [A, 98] # VAL98
- [A, 103] # PRO103
- [A, 104] # ALA104
- [A, 107] # SER107
- [A, 130] # HIS130
- [A, 131] # ARG131
- [A, 132] # LEU132
- [A, 181] # THR181
- [A, 184] # VAL184
- [A, 185] # VAL185
max_distance: 6.0
force: true
properties:
- affinity:
binder: Bpip install boltz
boltz predict job.yaml \
--use_msa_server \
--use_potentials \
--diffusion_samples 3 \
--recycling_steps 3 \
--step_scale 1.638Key result files:
| File | Contents |
|---|---|
*_model_0.pdb |
Predicted ternary structure |
confidence_*.json |
iptm, ptm, ligand_iptm |
affinity_*.json |
affinity_pred_value in kcal/mol |
pae_*.npz |
Predicted aligned error matrix |
Sanity checks:
iptm > 0.6— confident protein–protein interfaceligand_iptm > 0.5— confident ligand placement- A low off-diagonal block between chain
Aand chainCin the PAE map = confident interaction
v2.html— the second, broader tool. Live at https://ayushmania2002.github.io/boltzyml/v2.html.

Where v1 emits local-CLI YAML, v2.0 targets the hosted Boltz-2 API. It builds the
API payload (entities / templates / binding / model_options), repairs your template CIFs in the
browser, and submits jobs with your own API key. It is not limited to ternary complexes — define
any binary, ternary, or N-body mix of proteins, ligands (CCD or SMILES), DNA, and RNA.
| Stage | Capability |
|---|---|
| 1 · Build | Arbitrary entity sets (protein / ligand / DNA / RNA), per-chain IDs, binder selection, live residue/base counts, and sampling control matched to the API (num_samples 1–10, recycling_steps 1–10, sampling_steps ≥50, step_scale 1.3–2.0). Ligands by CCD code, SMILES, or a structure file (.sdf/.mol/.mol2) converted to SMILES in-browser via self-hosted RDKit. Per-protein MSA: automatic, single-sequence, or upload your own .a3m/.csv. |
| 1 · Clean templates | Drop a raw RCSB/ChimeraX .cif/.pdb. BoltzYML rewrites _struct_asym to remove phantom chains, strips waters/ligands, repairs modified residues (e.g. OCY → CYS), deletes _pdbx_poly_seq_scheme / _struct_conn records, and rebuilds polymer metadata for raw PDB files — the exact fixes that otherwise make the Boltz template parser fail. Then map each template chain to a prediction chain (one template per chain; duplicates are dropped). |
| 2 · Submit | Name the job (optional), estimate the exact cost via Boltz's estimate-cost endpoint before you commit, then paste your own Boltz API key and submit. No setup, no install. Your key lives only in your browser tab and is forwarded to Boltz through BoltzYML's open-source proxy, which stores nothing. |
| 3 · Results | Jobs poll themselves with a spinner (no over-clicking) and show the run time (from Boltz timestamps). View the structure in-browser (3Dmol viewer): colored by pLDDT confidence or chain, hover for residue name/number/chain, and drag-select residues from the sequence strip to highlight the interface in 3D. Read ipTM / pTM / pLDDT, and download a results .zip (every sample CIF + metrics.json + a branded README.txt) plus the best structure by ipTM. |
No installation, no proxy setup. The whole workflow is drop files → paste key → submit → download:
- Get a Boltz API key (instructions below).
- Open the app: https://ayushmania2002.github.io/boltzyml/v2.html
- Define your complex — add entities (protein sequence, ligand, DNA, RNA), give each a single-letter chain ID, and mark a ligand as binder if you want affinity/pose scoring. Sequence fields show a live amino-acid / base count.
- Ligands can be entered three ways: a CCD code (e.g.
A8S), a SMILES string, or an uploaded structure file (.sdf/.mol/.mol2). File ligands are converted to SMILES in your browser (self-hosted RDKit, ~7 MB loaded on first use); the result is shown in an editable field so you can verify it before submitting. - MSA (per protein): the default is server-generated (recommended). You can also choose single-sequence, or upload your own MSA (
.a3mor.csv) on any protein entity. The MSA field is protein-only, so in mixed complexes (protein + DNA/RNA/ligand) it simply applies to the protein chains.
- Ligands can be entered three ways: a CCD code (e.g.
- (Optional) Drop a template — a
.cif/.pdbfrom RCSB, AlphaFold, ChimeraX, etc. BoltzYML auto-cleans it in your browser (and converts legacy PDB to mmCIF, emitting the polymer metadata Boltz needs); then map each template chain to a prediction chain. Boltz allows one template per chain, so duplicate mappings are dropped automatically. - Set sampling options (or keep the defaults) and click Rebuild to preview the exact payload.
- (Optional) Name the job and click Estimate cost to get Boltz's exact USD estimate before committing.
- Paste your Boltz API key and click Submit prediction. Running jobs poll themselves with a spinner, so you do not need to keep clicking.
- Under Jobs & results, once
succeeded, click View 3D to see the predicted structure right in the page (colored by pLDDT confidence or by chain), or Download results — a.zipof every sample structure +metrics.json+README.txt, plus the best structure by ipTM. Files use your job name when set.
The hosted Boltz-2 API is a paid service, run by Boltz — separate from BoltzYML.
- Go to the Boltz API docs, create an account, and generate an API key.
- Boltz includes $5 of free credit every month; paid top-ups / subscriptions are available — check the current rates on the Boltz site/docs .
- Paste the key into BoltzYML v2.0. It stays in your browser tab only (optionally remembered for the session) and is never sent anywhere except through the proxy to Boltz.
Cost. Pricing is per sample (Boltz bills num_samples units), starting around $0.025–$0.05 per
sample and scaling with complex size, so a typical job is on the order of a few cents up to ~$0.50.
Rather than guess, BoltzYML has an Estimate cost button that calls Boltz's official estimate-cost
endpoint and shows the exact USD figure for your payload before you submit. Confirm current rates on the
Boltz site, since pricing can change.
Endpoints, pricing, and rate limits may change. Always check the official docs for current details.
All parsing and cleaning happens in your browser. Your raw files are never uploaded for processing.
- In your browser (client-side JavaScript):
- Sequences and ligand codes you type stay local.
- When you drop a template CIF/PDB, BoltzYML parses it locally and: detects which chains actually
have coordinates, rewrites
_struct_asymto drop phantom chains, strips waters/ligands (HETATM), maps modified residues (e.g.OCY → CYS) to standard parents, and removes_pdbx_poly_seq_scheme/_struct_connmetadata that would otherwise crash the Boltz parser. - It assembles the Boltz API payload (
entities/templates/binding/model_options).
- At submission, your browser sends the already-cleaned payload plus your API key to the proxy
— a small, stateless, open-source Cloudflare Worker (
worker/worker.js). The proxy:- uploads each cleaned template to transient storage so Boltz can fetch it by URL,
- forwards the payload to
api.boltz.biowith your key in a request header, - relays status polls and proxies result downloads (Boltz's result files are otherwise CORS-blocked for browsers).
Data handling / privacy:
- Your API key is forwarded to Boltz once per request and is never logged or stored by the proxy.
- Your sequences / payload pass through the proxy to Boltz and are not stored by the proxy.
- Cleaned template files are the only thing held, and only transiently — in a per-template Durable Object that auto-expires after 2 hours (Boltz needs a fetchable URL for templates).
- The proxy is stateless and open-source, so you can read exactly what it does.
api.boltz.bio sends no CORS headers, so a static page (GitHub Pages) can't call it directly, and result
files are likewise CORS-blocked. v2.0 therefore routes through a tiny stateless Cloudflare Worker. The
public app uses a hosted proxy maintained by the author — you don't deploy anything.
Prefer to run your own proxy? (e.g. for a lab, or to use your own free-tier quota.) Deploy the included Worker in a few minutes:
cd worker
npx wrangler login
npx wrangler deploy # Durable Object migrations create the stores automaticallyThen fork the app and change the hardcoded PROXY_URL constant in v2.html to your Worker's URL.
(By design there is no runtime ?proxy= override — see Security below.) See
worker/README.md. Templates are held in a strongly-consistent Durable Object
(readable globally the instant they are written, so Boltz's fetch never races a slow KV propagation) and
auto-expire after 2 hours. No payment method is required.
Because users supply their own (paid) Boltz API key, BoltzYML v2.0 has undergone an extensive security review focused on protecting that credential. The safeguards in place:
- Key transmission. The API key is sent only over TLS/HTTPS, and only in a request header
(
X-Boltz-Key) — never in a URL, query string, or request body. This keeps it out of browser history, server access logs,Refererheaders, and analytics. It is transmitted to exactly one origin: the hardcoded proxy. - Key storage. The key lives in volatile
sessionStorageonly when the user opts in, and is cleared when the tab closes. It is never written tolocalStorageand never leaves the browser except as above. - No runtime proxy override (anti-phishing). The proxy endpoint is a hardcoded constant with no
?proxy=/ query-string override. This deliberately removes a link-based key-exfiltration / open-redirect vector (a crafted?proxy=https://attacker.examplelink cannot retarget the key). - XSS / injection hardening. All dynamic or untrusted content — Boltz API responses, error messages, and
any text derived from uploaded files — is rendered via
textContent/ DOM text nodes, notinnerHTML, neutralizing reflected, stored, and DOM-based cross-site scripting. - Stateless, least-privilege proxy. The Cloudflare Worker holds no credentials of its own, forwards
each key to
api.boltz.bioexactly once, and never logs or persists it. - SSRF / open-proxy guard. The result-download endpoint (
/fetch) is restricted by a host allow-list (*.amazonaws.com,*.boltz.bio), so it cannot be abused to fetch arbitrary URLs. - Client-side processing. Template parsing and CIF cleaning run entirely in the browser; raw user files are never uploaded for processing.
- Transient, isolated storage. Cleaned templates live in per-template Durable Objects that auto-expire after 2 hours; the visit counter uses a separate Durable Object. No persistent user data is retained.
- Transport security. Served exclusively over HTTPS (GitHub Pages + Cloudflare TLS).
The proxy is open-source (worker/worker.js) so every one of these claims is
independently verifiable. Found a vulnerability? Please open a
security issue.
You don't need these to use BoltzYML (the web tool handles everything), but the Boltz API ships official client libraries if you want to script submissions directly:
| Language | Install | Docs |
|---|---|---|
TypeScript 0.43.0 |
npm install boltz-api |
docs |
Python 0.33.0 |
pip install boltz-api |
docs |
Go v0.23.0 |
go get -u 'github.com/boltz-bio/boltz-api-go@v0.0.1' |
docs |
CLI v0.28.0 |
irm https://install.boltz.bio/boltz-api/install.ps1 | iex |
docs |
boltzyml/
├── index.html # v1 web app — ternary CLI-YAML generator (GitHub Pages)
├── v2.html # v2.0 web app — hosted-API builder, template cleaner, submitter
├── vendor/rdkit/ # Self-hosted RDKit WASM — ligand file → SMILES, loaded on demand
├── vendor/3dmol/ # Self-hosted 3Dmol.js — in-browser structure viewer, loaded on demand
├── worker/ # Stateless Cloudflare Worker proxy for v2.0 submission
│ ├── worker.js # key passthrough + template hosting (Durable Object) + result fetch
│ ├── wrangler.toml # deploy config (Durable Object bindings)
│ └── README.md # one-time deploy instructions
├── boltzyml_v2_workflow.svg/.png # v2.0 workflow figure (site + README)
├── logo.png # Wordmark — favicon + header logo
├── banner.png # Pipeline schematic — used in this README
│
├── pyproject.toml # PyPI packaging metadata (hatchling)
├── LICENSE # MIT
│
├── src/boltzyml/
│ ├── __init__.py # Public API re-exports
│ ├── cli.py # CLI entry point (boltzyml command)
│ ├── parser.py # CIF parser (1-line and 2-line layouts)
│ ├── contacts.py # CA-to-ligand pocket contact computation
│ ├── utils.py # Chain assignment via k-mer similarity
│ └── yaml_writer.py # Boltz-2 v1 YAML emitter
│
└── tests/
├── test_parser.py # Synthetic CIFs for both layouts
├── test_contacts.py # Cutoff filtering, missing ligand, chain remap
└── test_cli.py # End-to-end CLI smoke test
pip install -e ".[dev]"
pytestOr run each file directly without pytest:
python tests/test_parser.py
python tests/test_contacts.py
python tests/test_cli.pyEach script exits non-zero on failure and prints all tests passed otherwise.
LIG/UNL/UNKligands. Docking tools often name the ligand generically. The real PDB CCD code (e.g.A8Sfor abscisic acid,ATP,HEM) belongs in theccd:field — use the CCD override in the web app or--ccdon the CLI. Verify codes at https://www.rcsb.org/ligand/.- Residue numbering. Boltz-2 uses
auth_seq_idfrom your CIF. If your CIF starts at residue 14 (truncated structure), the pocket numbers will start at 14 too — that is correct. - Apo vs holo Protein 1. For the ligand CIF, use the holo (ligand-bound) structure, not the apo AlphaFold prediction, so the pocket is in the right conformation.
- Tamarind Bio users. The same YAML works on https://app.tamarind.bio/boltz. Do not include a
templates:block when submitting there — use Tamarind's UI fields for template CIFs instead.
If you use BoltzYML in published or shared work, please cite both of the following.
1. BoltzYML (this tool):
Mallick, A. BoltzYML: input preparation and hosted-API submission tools for Boltz-2 structure and binding prediction. Zenodo (2026). Concept DOI (always latest): https://doi.org/10.5281/zenodo.20397272 · v2.0: https://doi.org/10.5281/zenodo.20440141
The concept DOI above always resolves to the newest release; cite a version DOI if you need to pin the exact version you used (v1: 10.5281/zenodo.20399843 · v2.0: 10.5281/zenodo.20440141).
BibTeX:
@software{boltzyml_2026,
author = {Mallick, Ayushman},
title = {{BoltzYML}: input preparation and hosted-API submission
tools for Boltz-2 structure and binding prediction},
year = {2026},
publisher = {Zenodo},
version = {2.0.0},
doi = {10.5281/zenodo.20397272},
url = {https://doi.org/10.5281/zenodo.20397272}
}2. Boltz-2 (the upstream model BoltzYML produces input for):
Wohlwend, J. et al. Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction. 2024. https://github.com/jwohlwend/boltz
If you use the ligand-file → SMILES conversion, also acknowledge RDKit (see below).
- RDKit — ligand structure files (
.sdf/.mol/.mol2) are converted to SMILES in the browser using RDKit (RDKit.js /@rdkit/rdkit), redistributed invendor/rdkit/under the BSD 3-Clause License (Copyright © Valence Discovery Inc., Greg Landrum, Paolo Tosco, and other RDKit contributors). Seevendor/rdkit/LICENSE. RDKit: Open-source cheminformatics. https://www.rdkit.org. - 3Dmol.js — the in-browser structure viewer (pLDDT / chain coloring) uses 3Dmol.js, redistributed in
vendor/3dmol/under the BSD 3-Clause License. Seevendor/3dmol/LICENSE. Rego & Koes, 3Dmol.js: molecular visualization with WebGL, Bioinformatics (2015). - Boltz — for the Boltz-2 models and the hosted API.
MIT. Copyright © 2026 Ayushman Mallick. Bundled third-party components retain their own licenses (e.g. RDKit, BSD 3-Clause, in vendor/rdkit/LICENSE).