

LaminDB is an open-source data framework for biology to query, trace, and validate datasets and models at scale. You get context & memory through a lineage-native lakehouse that understands bio-formats, registries & ontologies.
Why?
(1) Reproducing, tracing & understanding how datasets, models & results are created is critical to quality R&D. Without context, humans & agents make mistakes and cannot close feedback loops across data generation & analysis. Without memory, compute & intelligence are wasted on fragmented, non-compounding tasks.
(2) Training & fine-tuning models with thousands of datasets — across LIMS, ELNs, orthogonal assays — is now a primary path to scaling R&D. But without queryable & validated data or with data locked in organizational & infrastructure siloes, it leads to garbage in, garbage out or is quite simply impossible.
Imagine building software without git or pull requests: an agent's quality would be impossible to verify. While code has git and tables have dbt/warehouses, biological data has lacked a framework for managing its unique complexity.
LaminDB fills the gap.
It is a lineage-native lakehouse that understands bio-registries and formats (AnnData, .zarr, …) based on the established open data stack:
Postgres/SQLite for metadata and cross-platform storage for datasets.
By offering queries, tracing & validation in a single API, LaminDB provides the context & memory to turn messy, agentic biological R&D into a scalable process.

How?
- lineage → track inputs & outputs of notebooks, scripts, functions & pipelines with a single line of code
- lakehouse → manage, monitor & validate schemas for standard and bio formats; query across many datasets
- FAIR datasets → validate & annotate
DataFrame,AnnData,SpatialData,parquet,zarr, … - LIMS & ELN → programmatic experimental design with bio-registries, ontologies & markdown notes
- unified access → storage locations (local, S3, GCP, …), SQL databases (Postgres, SQLite) & ontologies
- reproducible → auto-track source code & compute environments with data & code versioning
- change management → branching & merging similar to git
- zero lock-in → runs anywhere on open standards (Postgres, SQLite,
parquet,zarr, etc.) - scalable → you hit storage & database directly through your
pydataor R stack, no REST API involved - simple → just
pip installfrom PyPI orinstall.packages('laminr')from CRAN - distributed → zero-copy & lineage-aware data sharing across infrastructure (databases & storage locations)
- integrations → git, nextflow, vitessce, redun, and more
- extensible → create custom plug-ins based on the Django ORM, the basis for LaminDB's registries
GUI, permissions, audit logs? LaminHub is a collaboration hub built on LaminDB similar to how GitHub is built on git.
Who?
Scientists and engineers at leading research institutions and biotech companies, including:
- Industry → Pfizer, Altos Labs, Ensocell Therapeutics, ...
- Academia & Research → scverse, DZNE (National Research Center for Neuro-Degenerative Diseases), Helmholtz Munich (National Research Center for Environmental Health), ...
- Research Hospitals → Global Immunological Swarm Learning Network: Harvard, MIT, Stanford, ETH Zürich, Charité, U Bonn, Mount Sinai, ...
From personal research projects to pharma-scale deployments managing petabytes of data across:
| entities | OOMs |
|---|---|
| observations & datasets | 10¹² & 10⁶ |
| runs & transforms | 10⁹ & 10⁵ |
| proteins & genes | 10⁹ & 10⁶ |
| biosamples & species | 10⁵ & 10² |
| ... | ... |
Copy llms.txt into an LLM chat and let AI explain or read the docs.
Install the Python package:
pip install lamindbYou can browse public databases at lamin.ai/explore. To query laminlabs/cellxgene, run:
import lamindb as ln
db = ln.DB("laminlabs/cellxgene") # a database object for queries
df = db.Artifact.to_dataframe() # a dataframe listing datasets & modelsTo get a specific dataset, run:
artifact = db.Artifact.get("BnMwC3KZz0BuKftR") # a metadata object for a dataset
artifact.describe() # describe the context of the datasetSee the output.
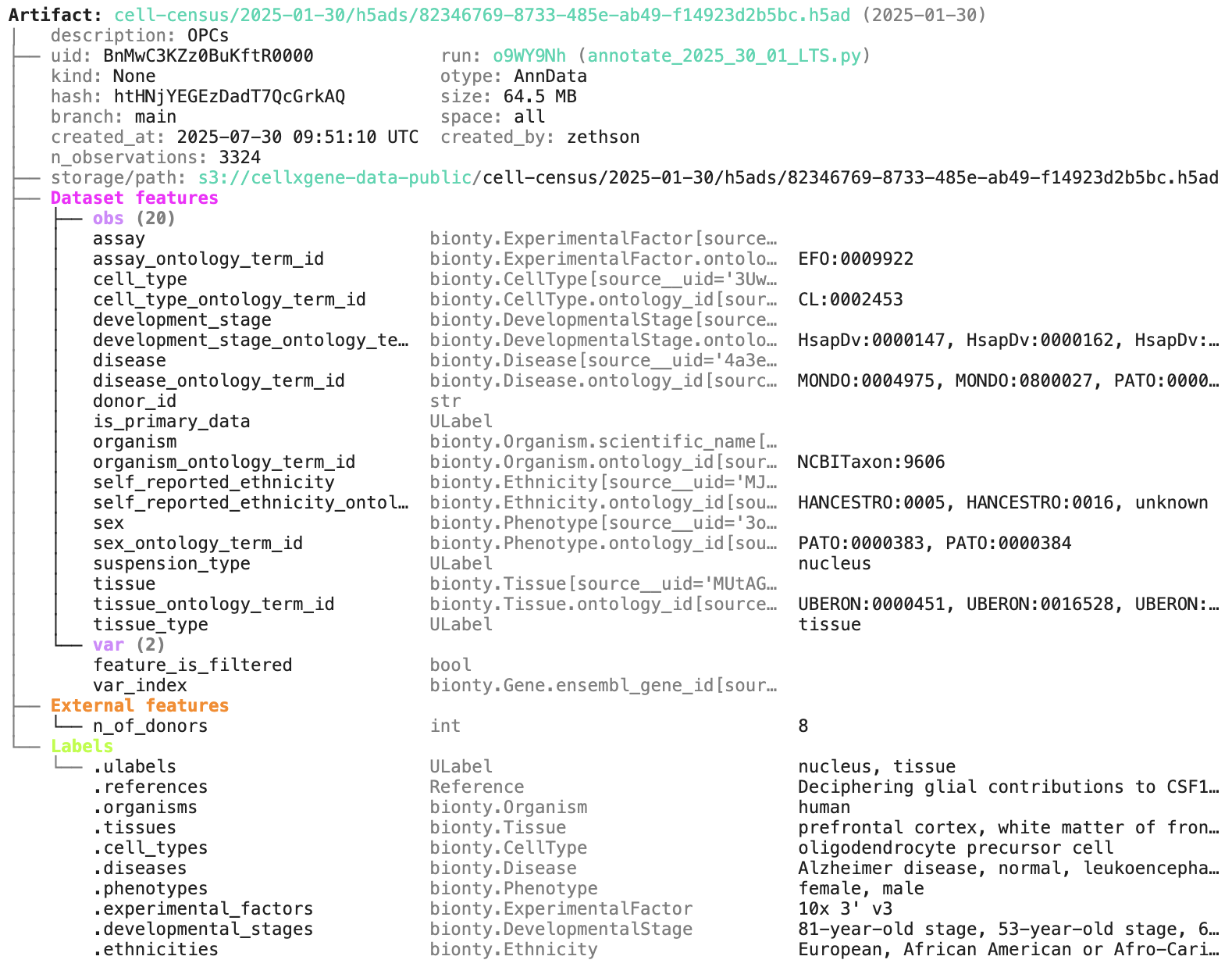
Access the content of the dataset via:
local_path = artifact.cache() # return a local path from a cache
adata = artifact.load() # load object into memory
accessor = artifact.open() # return a streaming accessorYou can query by biological entities like Disease through plug-in bionty:
alzheimers = db.bionty.Disease.get(name="Alzheimer disease")
df = db.Artifact.filter(diseases=alzheimers).to_dataframe()You can create a LaminDB instance at lamin.ai and invite collaborators. To connect to a remote instance, run:
lamin login
lamin connect account/nameIf you prefer to work with a local SQLite database (no login required), run this instead:
lamin init --storage ./quickstart-data --modules biontyOn the terminal and in a Python session, LaminDB will now auto-connect.
To save a file or folder from the command line, run:
lamin save myfile.txt --key examples/myfile.txtTo sync a file into a local cache (artifacts) or development directory (transforms), run:
lamin load --key examples/myfile.txtRead more: docs.lamin.ai/cli.
To create a dataset while tracking source code, inputs, outputs, logs, and environment:
import lamindb as ln
# → connected lamindb: account/instance
ln.track() # track code execution
open("sample.fasta", "w").write(">seq1\nACGT\n") # create dataset
ln.Artifact("sample.fasta", key="sample.fasta").save() # save dataset
ln.finish() # mark run as finishedRunning this snippet as a script (python create-fasta.py) produces the following data lineage:
artifact = ln.Artifact.get(key="sample.fasta") # get artifact by key
artifact.describe() # context of the artifact
artifact.view_lineage() # fine-grained lineage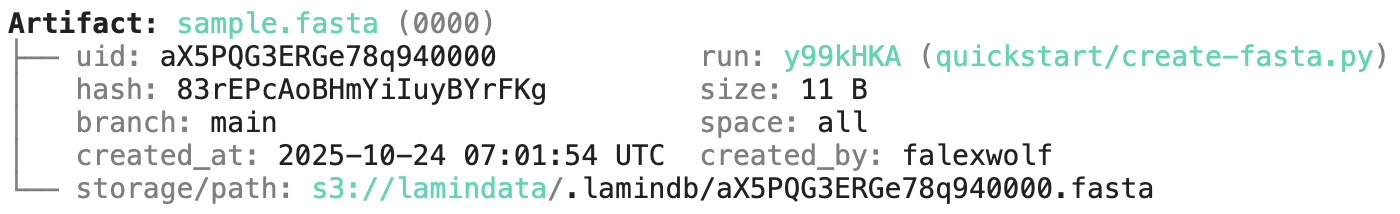
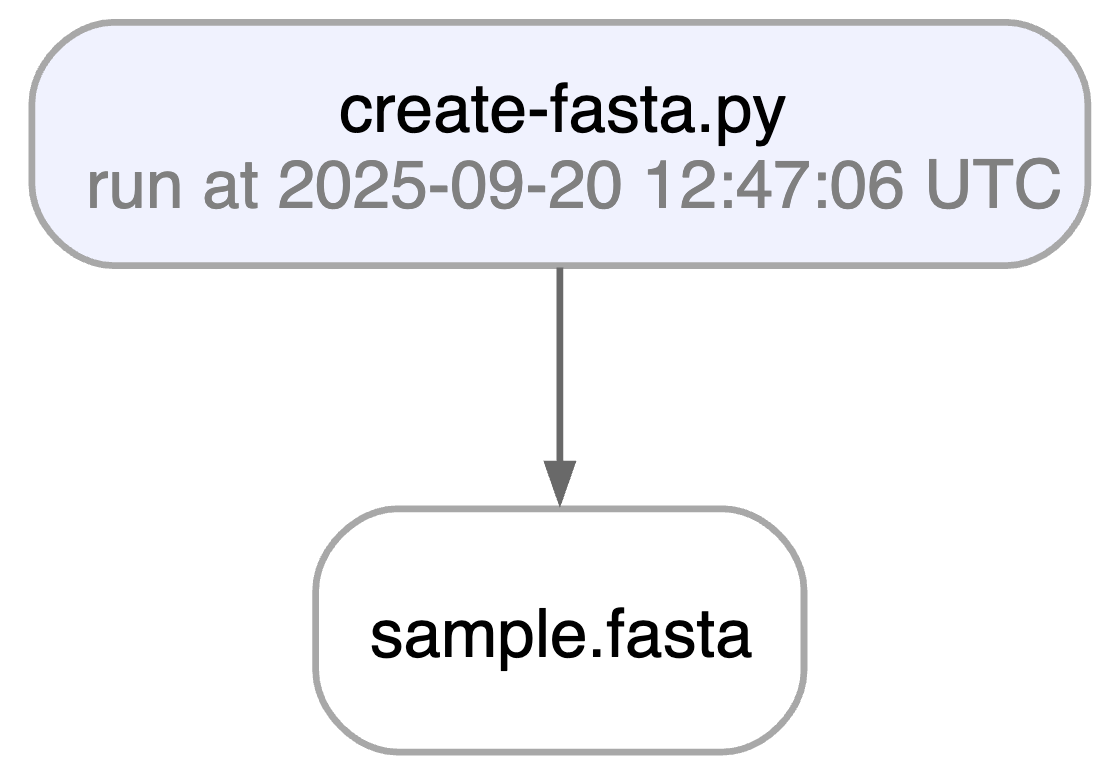
Access run & transform.
run = artifact.run # get the run object
transform = artifact.transform # get the transform object
run.describe() # context of the run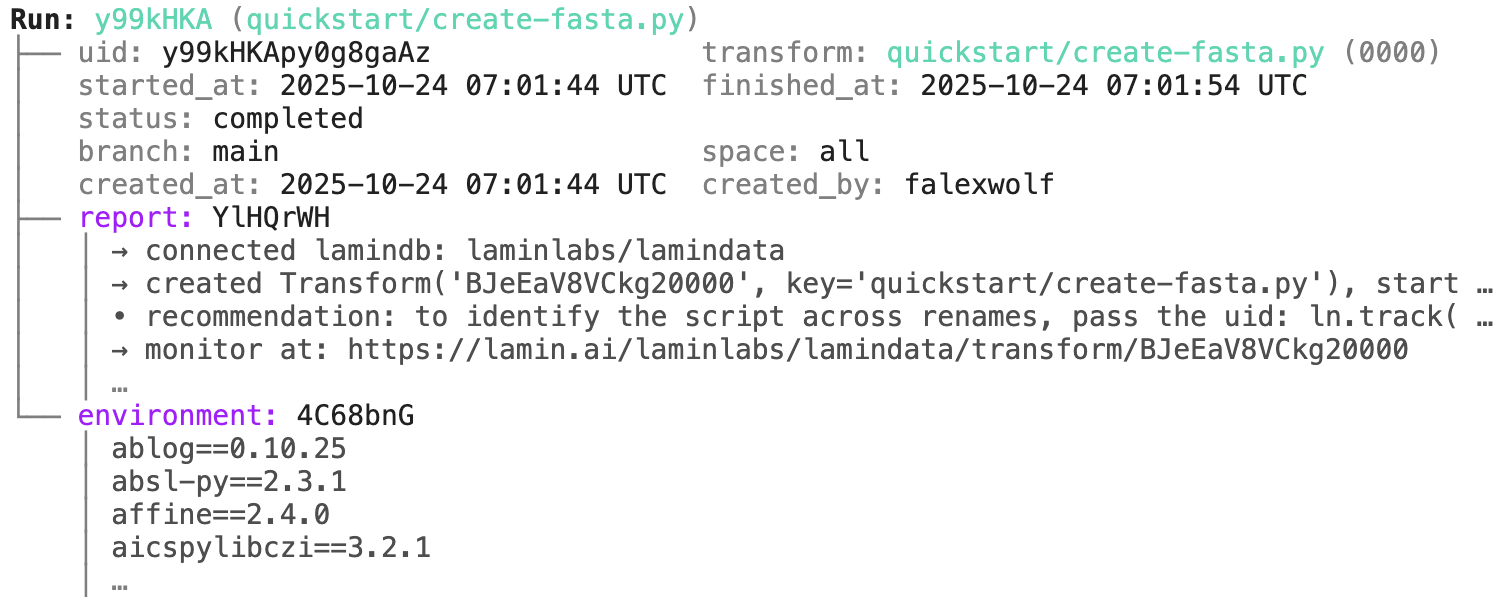
transform.describe() # context of the transform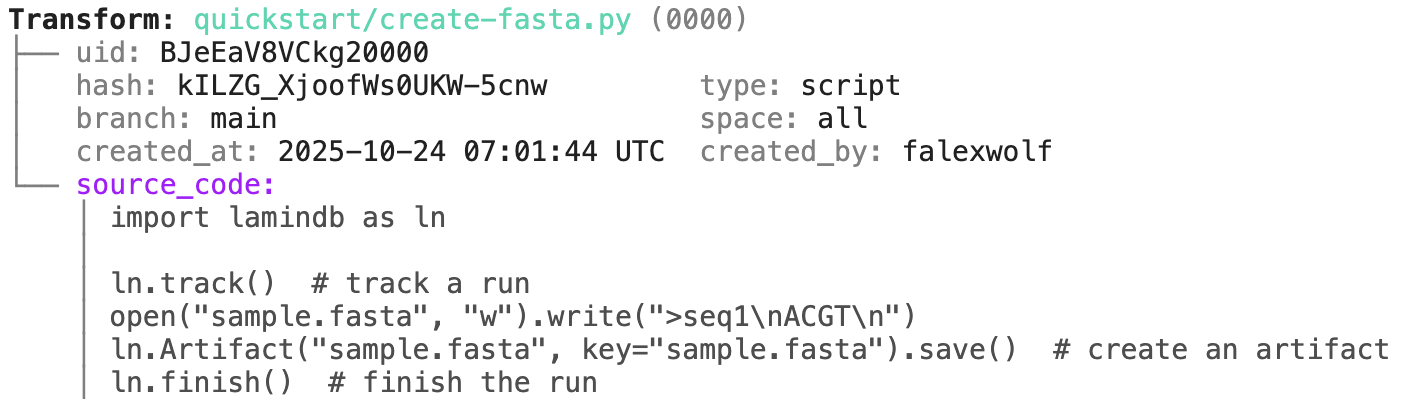
You can achieve the same traceability for functions & workflows:
import lamindb as ln
@ln.flow()
def create_fasta(fasta_file: str = "sample.fasta"):
open(fasta_file, "w").write(">seq1\nACGT\n") # create dataset
ln.Artifact(fasta_file, key=fasta_file).save() # save dataset
if __name__ == "__main__":
create_fasta()Beyond what you get for scripts & notebooks, this automatically tracks function & CLI params and integrates well with established Python workflow managers: docs.lamin.ai/track. To integrate advanced bioinformatics pipeline managers like Nextflow, see docs.lamin.ai/pipelines.
A richer example.
Here is a an automatically generated re-construction of the project of Schmidt el al. (Science, 2022):
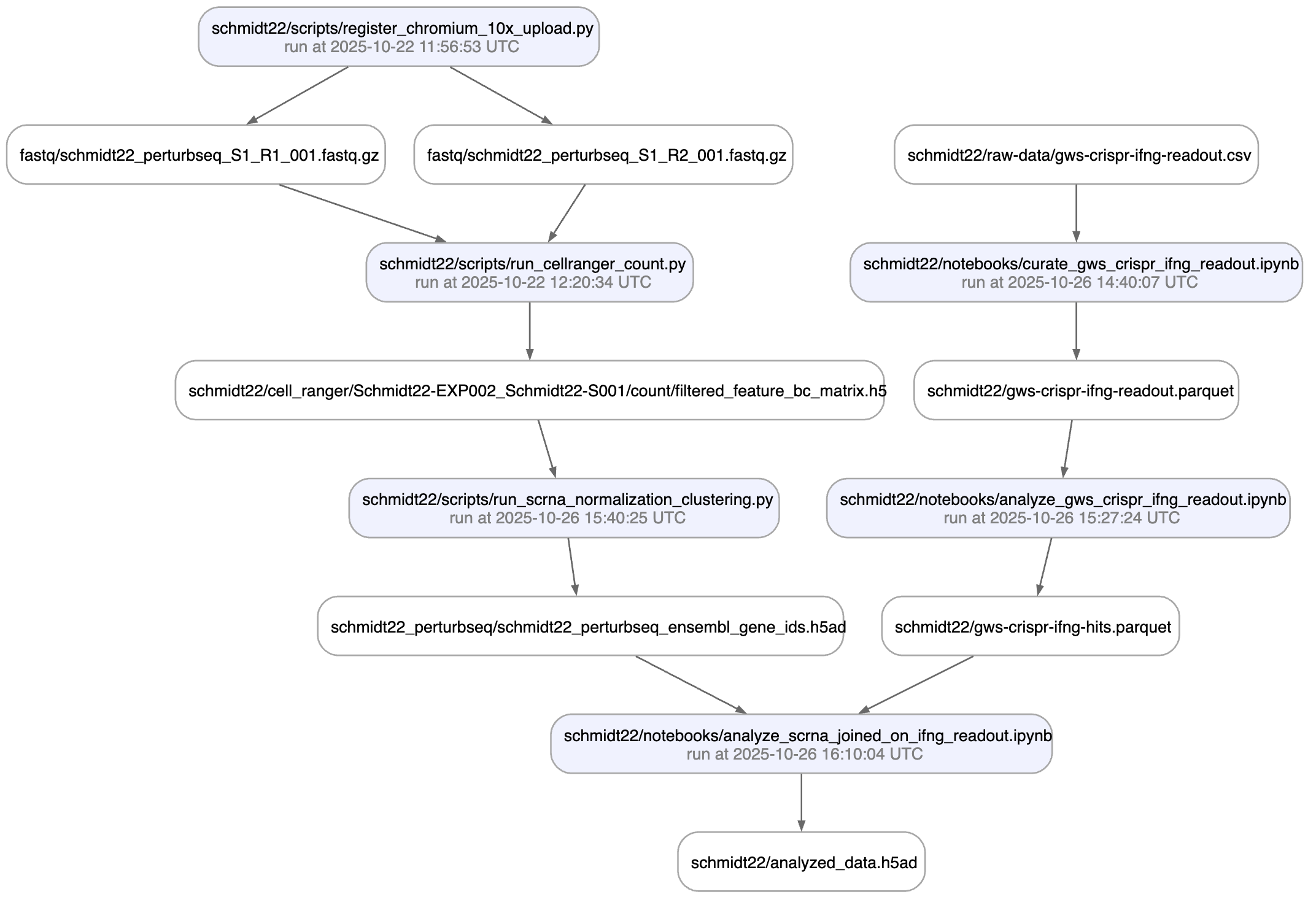
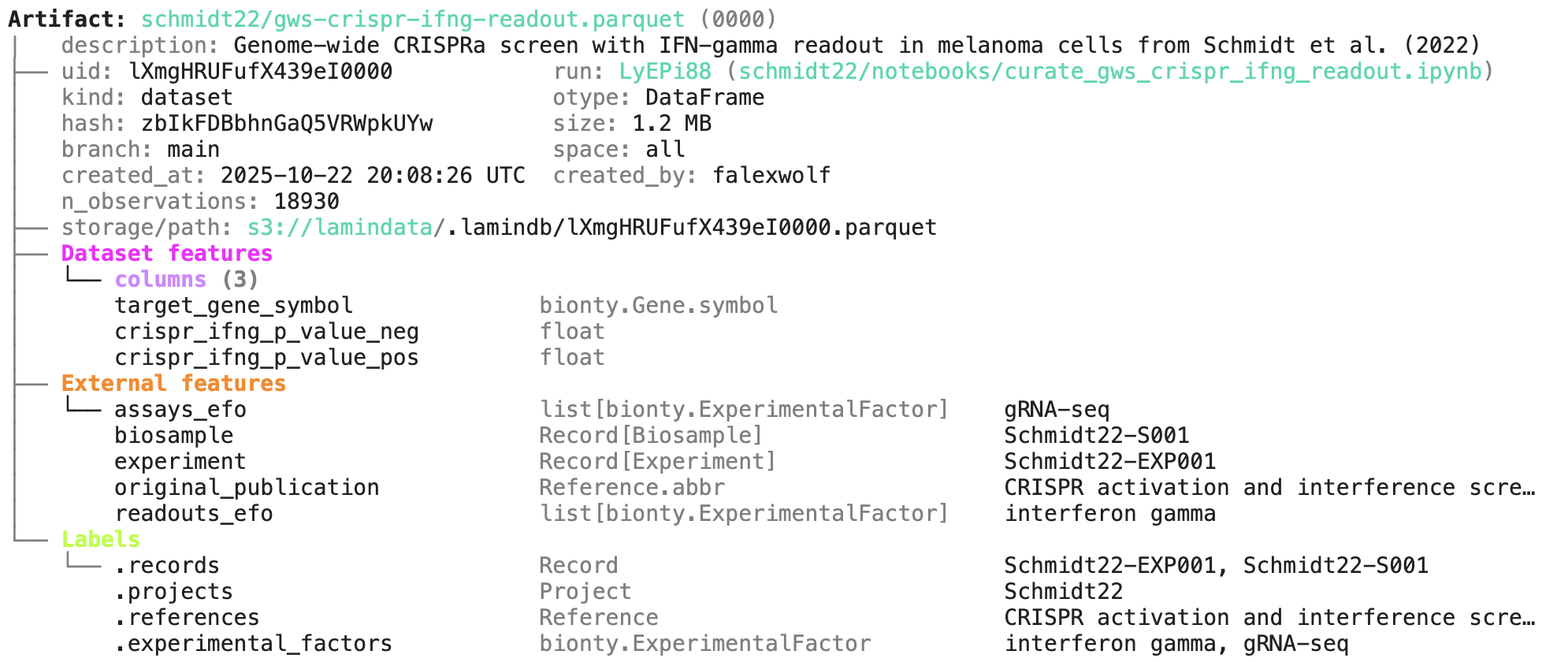
A phenotypic CRISPRa screening result is integrated with scRNA-seq data. Here is the result of the screen input:
You can label an artifact by running:
my_label = ln.ULabel(name="My label").save() # a universal label
project = ln.Project(name="My project").save() # a project label
artifact.ulabels.add(my_label)
artifact.projects.add(project)Query for it:
ln.Artifact.filter(ulabels=my_label, projects=project).to_dataframe()You can also query by the metadata that lamindb automatically collects:
ln.Artifact.filter(run=run).to_dataframe() # by creating run
ln.Artifact.filter(transform=transform).to_dataframe() # by creating transform
ln.Artifact.filter(size__gt=1e6).to_dataframe() # size greater than 1MBIf you want to include more information into the resulting dataframe, pass include.
ln.Artifact.to_dataframe(include=["created_by__name", "storage__root"]) # include fields from related registriesNote: The query syntax for DB objects and for your default database is the same.
You can annotate datasets and samples with features. Let's define some:
from datetime import date
ln.Feature(name="gc_content", dtype=float).save()
ln.Feature(name="experiment_note", dtype=str).save()
ln.Feature(name="experiment_date", dtype=date, coerce=True).save() # accept date stringsDuring annotation, feature names and data types are validated against these definitions:
artifact.features.add_values({
"gc_content": 0.55,
"experiment_note": "Looks great",
"experiment_date": "2025-10-24",
})Query for it:
ln.Artifact.filter(experiment_date="2025-10-24").to_dataframe() # query all artifacts annotated with `experiment_date`If you want to include the feature values into the dataframe, pass include.
ln.Artifact.to_dataframe(include="features") # include the feature annotationsYou can create records for the entities underlying your experiments: samples, perturbations, instruments, etc., for example:
sample = ln.Record(name="Sample", is_type=True).save() # create entity type: Sample
ln.Record(name="P53mutant1", type=sample).save() # sample 1
ln.Record(name="P53mutant2", type=sample).save() # sample 2Define features and annotate an artifact with a sample:
ln.Feature(name="design_sample", dtype=sample).save()
artifact.features.add_values({"design_sample": "P53mutant1"})You can query & search the Record registry in the same way as Artifact or Run.
ln.Record.search("p53").to_dataframe()You can also create relationships of entities and edit them like Excel sheets in a GUI via LaminHub.
If you change source code or datasets, LaminDB manages versioning for you.
Assume you run a new version of our create-fasta.py script to create a new version of sample.fasta.
import lamindb as ln
ln.track()
open("sample.fasta", "w").write(">seq1\nTGCA\n") # a new sequence
ln.Artifact("sample.fasta", key="sample.fasta", features={"design_sample": "P53mutant1"}).save() # annotate with the new sample
ln.finish()If you now query by key, you'll get the latest version of this artifact with the latest version of the source code linked with previous versions of artifact and source code are easily queryable:
artifact = ln.Artifact.get(key="sample.fasta") # get artifact by key
artifact.versions.to_dataframe() # see all versions of that artifactHere is how you ingest a DataFrame:
import pandas as pd
df = pd.DataFrame({
"sequence_str": ["ACGT", "TGCA"],
"gc_content": [0.55, 0.54],
"experiment_note": ["Looks great", "Ok"],
"experiment_date": [date(2025, 10, 24), date(2025, 10, 25)],
})
ln.Artifact.from_dataframe(df, key="my_datasets/sequences.parquet").save() # no validationTo validate & annotate the content of the dataframe, use the built-in schema valid_features:
ln.Feature(name="sequence_str", dtype=str).save() # define a remaining feature
artifact = ln.Artifact.from_dataframe(
df,
key="my_datasets/sequences.parquet",
schema="valid_features" # validate columns against features
).save()
artifact.describe()You can filter for datasets by schema and then launch distributed queries and batch loading.
To validate an AnnData with built-in schema ensembl_gene_ids_and_valid_features_in_obs, call:
import anndata as ad
import numpy as np
adata = ad.AnnData(
X=pd.DataFrame([[1]*10]*21).values,
obs=pd.DataFrame({'cell_type_by_model': ['T cell', 'B cell', 'NK cell'] * 7}),
var=pd.DataFrame(index=[f'ENSG{i:011d}' for i in range(10)])
)
artifact = ln.Artifact.from_anndata(
adata,
key="my_datasets/scrna.h5ad",
schema="ensembl_gene_ids_and_valid_features_in_obs"
)
artifact.describe()To validate a spatialdata or any other array-like dataset, you need to construct a Schema. You can do this by composing simple pandera-style schemas: docs.lamin.ai/curate.
Plugin bionty gives you >20 public ontologies as SQLRecord registries. This was used to validate the ENSG ids in the adata just before.
import bionty as bt
bt.CellType.import_source() # import the default ontology
bt.CellType.to_dataframe() # your extendable cell type ontology in a simple registryRead more: docs.lamin.ai/manage-ontologies.