
| Overview: | Assemble long reads (PacBio HiFi, PacBio subreads, Nanopore) into high-quality genome assemblies with optional polishing, annotation, and quality assessment. |
|---|---|
| Input: | BAM files from PacBio sequencers, or FastQ files from Nanopore or PacBio HiFi sequencers. |
| Output: | HTML reports with per-sample assembly statistics, coverage, BLAST identification, BUSCO scores, and optional annotation. |
| Status: | Production |
| Citation: |
|
pip install sequana-lora
To upgrade an existing installation:
pip install sequana-lora --upgrade
Step 1 — prepare the working directory:
sequana_lora \
--input-directory /path/to/reads \
--data-type pacbio-hifi \
--assembler flye \
--genome-size 3m \
--apptainer-prefix /path/to/containers
This creates a lora/ working directory containing config.yaml and a
lora.sh launch script.
Step 2 — review the configuration (optional but recommended):
cd lora cat config.yaml # adjust parameters as needed
Step 3 — run the pipeline:
sh lora.sh
Or launch directly from step 1 with --execute (skips the review step):
sequana_lora ... --execute
To watch live progress in the terminal, add --monitor:
sequana_lora ... --execute --monitor
Three options are always required:
--assembler- Assembler to use. Choices:
flye(recommended for HiFi),canu,hifiasm,unicycler,necat,pecat. --data-typeTechnology and quality of the input reads:
Value Description pacbio-hifi PacBio HiFi / CCS reads (Q20+) pacbio-raw PacBio CLR / subreads (raw) pacbio-corr PacBio corrected reads nano-hq Nanopore Q20+ reads (e.g. R10.4 with SUP basecalling) nano-raw Nanopore standard reads nano-corr Nanopore corrected reads --genome-size- Estimated genome size, e.g.
3m(3 Mb),2.5g(2.5 Gb). Required by Flye; used by Canu for coverage reporting.
sequana_lora \
--input-directory /data/hifi \
--data-type pacbio-hifi \
--assembler flye \
--genome-size 3m \
--apptainer-prefix /shared/containers \
--do-coverage
Use --mode bacteria to enable sequana_coverage, prokka, busco, and checkm
in one shot:
sequana_lora \
--input-directory /data/nanopore \
--data-type nano-hq \
--assembler flye \
--genome-size 3m \
--apptainer-prefix /shared/containers \
--mode bacteria \
--busco-lineage bacteria \
--checkm-rank genus \
--checkm-name Streptococcus
If your input is raw PacBio BAM files (subreads), LORA can build CCS/HiFi reads first:
sequana_lora \
--input-directory /data/subreads \
--data-type pacbio-raw \
--assembler flye \
--genome-size 3m \
--pacbio-build-ccs \
--pacbio-ccs-min-passes 10 \
--pacbio-ccs-min-rq 0.99
Or, if you have multiple BAM files per sample, provide a CSV:
sequana_lora \
--pacbio-input-csv samples.csv \
--data-type pacbio-raw \
--assembler flye \
--genome-size 3m
The CSV format is: one row per sample with columns sample,file1[,file2,...].
Computes depth of coverage and breadth of coverage for each contig using sequana_coverage. Highly recommended to check assembly quality:
--do-coverage
Assess genome completeness against a lineage-specific marker gene set:
--busco-lineage bacteria # auto-download bacteria lineage --busco-lineage streptococcales # specific clade --busco-print-lineages # list all available lineages
Annotate contigs (bacterial genomes):
--do-prokka
Estimate completeness and contamination for bacterial genomes:
--checkm-rank genus --checkm-name Streptococcus
Use an invalid --checkm-name value to get a list of valid names for a
given rank, e.g. --checkm-rank genus --checkm-name HELP.
Polish long-read contigs with paired-end Illumina data:
--do-polypolish \ --polypolish-input-directory /data/illumina \ --polypolish-input-pattern "*.fastq.gz" \ --polypolish-input-readtag "_R[12]_"
Explicit circularisation with Circlator (Flye performs this automatically):
--do-circlator
BLAST aligns each contig against a nucleotide database to identify the assembled sequences. The top hits appear in the HTML report.
Requires a locally installed BLAST+ and a downloaded nt database
(~270 GB). Fastest option with no network dependency:
--blastdb /path/to/blast/databases
No local database required — jobs are submitted to NCBI's BLAST servers. Enable by providing an email address:
--blast-email your@email.com
Jobs are submitted sequentially (one contig at a time) to avoid IP-level CPU
throttling by NCBI. The default database is nt; use --blast-remote-db
to change it:
--blast-email your@email.com --blast-remote-db refseq_genomic
Restricting the search to an organism group (strongly recommended)
Searching all of nt for a large contig is slow and prone to NCBI CPU
throttling. Restrict the search by editing config.yaml after running
sequana_lora. The entrez_query parameter is equivalent to filling the
"Organism" box on the NCBI BLAST web form.
Option 1 — curated bacterial reference genomes (fastest, recommended for bacteria)
Use refseq_genomic as the database and restrict to bacteria. RefSeq contains
only complete, curated reference genomes — a much smaller and higher-quality
search space than nt:
blast:
remote_db: 'refseq_genomic'
entrez_query: 'Bacteria[Organism]'
Option 2 — all bacteria in nt, reference sequences only
Stay on nt but filter to RefSeq-quality entries:
blast:
remote_db: 'nt'
entrez_query: 'Bacteria[Organism] AND refseq[filter]'
Option 3 — single genus
Useful when the organism is known:
blast:
remote_db: 'nt'
entrez_query: 'Streptococcus[Organism]'
Note
If your organism is novel or not yet in RefSeq, fall back to
remote_db: nt with a broad entrez_query such as
Bacteria[Organism].
NCBI API key (optional but recommended)
Register for a free NCBI API key at https://www.ncbi.nlm.nih.gov/account/
(sign in → API Key Management). It raises the rate limit from 3 to 10
requests/second and reduces CPU throttling for large queries. Add it to
config.yaml:
blast:
api_key: 'YOUR_KEY_HERE'
On a cluster with SLURM, pass --profile slurm:
sequana_lora \
--input-directory /data/hifi \
--data-type pacbio-hifi \
--assembler flye \
--genome-size 3m \
--profile slurm \
--slurm-queue fast \
--jobs 40 \
--apptainer-prefix /shared/containers
Per-rule memory and thread settings are controlled via the resources blocks
in config.yaml.
Every tool runs inside a pre-built container. Point --apptainer-prefix to a
shared directory so images are downloaded once and reused across projects:
--apptainer-prefix /shared/containers
Images are downloaded automatically on first run from Zenodo. Pass extra bind
mounts with --apptainer-args if your data lives outside $HOME:
--apptainer-args "-B /data:/data"
After running sequana_lora, a config.yaml is created in the working
directory. All pipeline parameters can be tuned there. Key sections:
assembler— which assembler to useflye/canu/hifiasm— assembler-specific optionsfastp— read filtering (minimum length, etc.)blast— BLAST settings includingentrez_queryandapi_keybusco/prokka/checkm— optional QC toolssequana_coverage— coverage analysis parametersmultiqc— aggregated report settings
Full reference: config.yaml
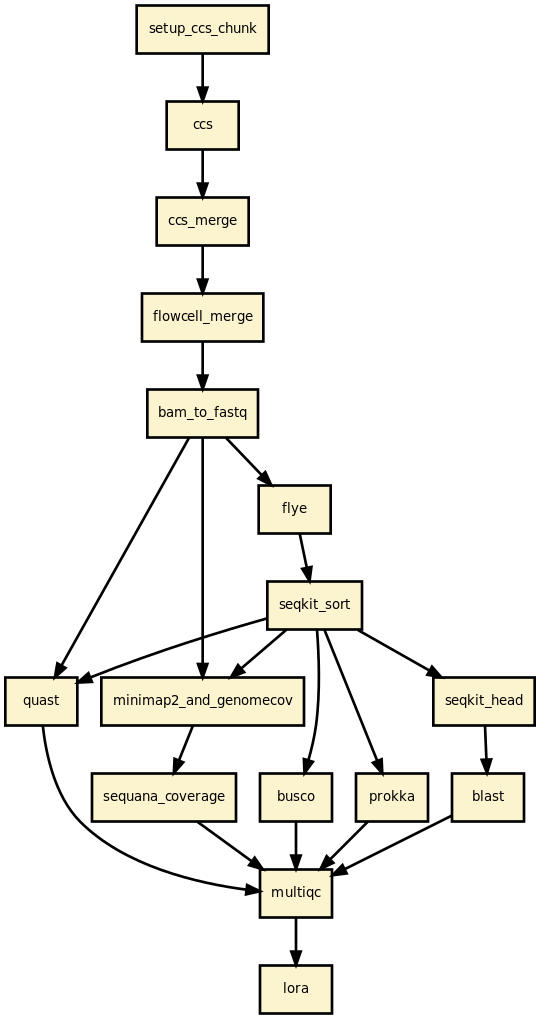
- Read filtering — fastp removes reads below the minimum length threshold.
- [Optional] CCS — build HiFi reads from PacBio subreads (ccs tool).
- Assembly — Flye / Canu / Hifiasm / Unicycler / NECAT / PECAT.
- [Optional] Circularisation — Circlator (or built into Flye).
- [Optional] Polishing — Polypolish with paired-end Illumina reads.
- Contig sorting — SeqKit sorts contigs by length (largest first).
- Read mapping — Minimap2 maps reads back to contigs; Mosdepth computes coverage.
- [Optional] Coverage analysis — sequana_coverage per contig.
- Quality assessment — QUAST assembly statistics.
- [Optional] BLAST — top hits per contig (local or remote NCBI).
- [Optional] BUSCO — genome completeness.
- [Optional] Prokka — genome annotation.
- [Optional] CheckM — contamination and completeness for bacteria.
- Reports — per-sample HTML report and a multi-sample summary.
| Version | Description |
|---|---|
| 1.1.0 |
|
| 1.0.0 |
|
| 0.3.0 |
|
| 0.2.0 |
|
| 0.1.0 | First release. |