
This is the mapper pipeline from the Sequana projet
| Overview: | This is a simple pipeline to map several FastQ files onto a reference using different mappers/aligners |
|---|---|
| Input: | A set of FastQ files (illumina, pacbio, etc). |
| Output: | A set of BAM files (and/or bigwig) and HTML report |
| Status: | Production |
| Documentation: | This README file, and https://sequana.readthedocs.io |
| Citation: | Cokelaer et al, (2017), 'Sequana': a Set of Snakemake NGS pipelines, Journal of Open Source Software, 2(16), 352, JOSS DOI https://doi:10.21105/joss.00352 |
If you already have all requirements, you can install the packages using pip:
pip install sequana_mapper --upgrade
You will need third-party software such as fastqc. Please see below for details.
Scan FastQ files in a directory and set up the pipeline (replace DATAPATH and genome.fa with your inputs):
sequana_mapper --input-directory DATAPATH --reference-file genome.fa --aligner-choice bwa sequana_mapper --input-directory DATAPATH --reference-file genome.fa --aligner-choice bwa --do-coverage sequana_mapper --input-directory DATAPATH --reference-file genome.fa --aligner-choice bwa --create-bigwig
For long-read data, use the dedicated presets:
sequana_mapper --input-directory DATAPATH --reference-file genome.fa --pacbio # sets minimap2 -x map-pb sequana_mapper --input-directory DATAPATH --reference-file genome.fa --nanopore # sets minimap2 -x map-ont
For capture-seq projects (feature counting):
sequana_mapper --input-directory DATAPATH --reference-file genome.fa --capture-annotation-file targets.saf
This creates a mapper/ directory with the pipeline and configuration file. Execute the pipeline locally:
cd mapper sh mapper.sh
See .sequana/profile/config.yaml to tune Snakemake behaviour (cores, cluster settings, etc.).
With apptainer, initiate the working directory as follows:
sequana_mapper --input-directory DATAPATH --reference-file genome.fa --use-apptainer
Images are downloaded in the working directory but you can store them in a shared location:
sequana_mapper --input-directory DATAPATH --reference-file genome.fa --use-apptainer --apptainer-prefix ~/.sequana/apptainers
and then:
cd mapper sh mapper.sh
This pipeline requires the following executables (install via bioconda/conda):
- bwa — short-read aligner (default)
- minimap2 — long-read aligner (PacBio / Nanopore)
- bowtie2 — alternative short-read aligner
- samtools / sambamba — BAM processing
- bamtools — BAM statistics
- deeptools — bigwig generation (
bamCoverage) - bedtools — genome arithmetic
- subread — feature counting (
featureCounts, capture-seq only) - mosdepth — fast coverage depth
- seqkit — FASTQ statistics
- multiqc — aggregated HTML report
- sequana_coverage — coverage analysis (prokaryotes)
Install all dependencies at once:
mamba env create -f environment.yml
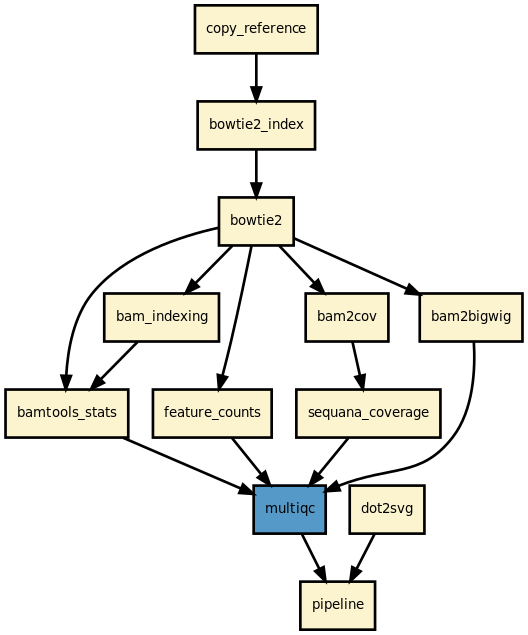
This pipeline maps FastQ files (paired or single-end) in parallel onto a reference genome and produces filtered BAM files, a MultiQC HTML report, and optionally coverage tracks and feature counts.
Aligner choice (--aligner-choice):
bwa(default) — BWA-MEM; index algorithm is auto-selected (isorbwtsw) based on reference sizebwa_split— experimental; splits large FastQs into 1 M-read chunks for parallel BWA jobs, then mergesminimap2— long-read aligner; use--pacbio(sets-x map-pb) or--nanopore(sets-x map-ont)bowtie2— standard short-read aligner
BAM filtering: unmapped reads are removed to minimise file size. Statistics reported by MultiQC
(in {sample}/bamtools_stats/) still include both mapped and unmapped read counts.
Optional outputs:
--do-coverage— runssequana_coveragefor depth-of-coverage analysis (prokaryotes)--create-bigwig— generates bigwig files viabamCoverage(deeptools)--capture-annotation-file— enablesfeatureCountsfor capture-seq efficiency metrics
Here is the latest documented configuration file to be used with the pipeline. Each rule used in the pipeline may have a section in the configuration file.
| Version | Description |
|---|---|
| 1.4.1 |
|
| 1.4.0 |
|
| 1.3.1 |
|
| 1.3.0 |
|
| 1.2.1 |
|
| 1.2.0 |
|
| 1.1.0 |
|
| 1.0.0 |
|
| 0.12.0 |
|
| 0.11.1 |
|
| 0.11.0 |
|
| 0.10.1 |
|
| 0.10.0 |
|
| 0.9.0 |
|
| 0.8.13 |
|
| 0.8.12 |
|
| 0.8.11 |
|
| 0.8.10 |
|
| 0.8.9 |
|
| 0.8.8 |
|
| 0.8.7 |
|
| 0.8.6 |
|
| 0.8.5 |
|
| 0.8.4 |
|
| 0.8.3 |
|
| 0.8.2 |
|
| 0.8.1 |
|
| 0.8.0 | First release. |
To contribute to this project, please take a look at the Contributing Guidelines first. Please note that this project is released with a Code of Conduct. By contributing to this project, you agree to abide by its terms.